Trois questions à François Dehez
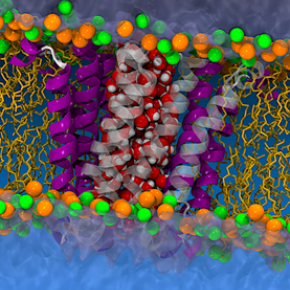
Avec ses collègues du Laboratoire de physique et chimie théoriques (LPCT, CNRS/Université de Lorraine), tels que Christophe Chipot, et de l’université d’Oxford, François Dehez souligne les problèmes d’identification des structures de protéines membranaires dans une publication de la revue Nature. Son exemple est basé sur la protéine p7, impliquée dans l’hépatite C.
Que sont les protéines membranaires et pourquoi intéressent-elles tant la recherche ?
Les protéines membranaires sont enchâssées dans les membranes biologiques, où elles assurent la communication entre l’intérieur et l’extérieur des cellules : ions, molécules, signaux, etc. Ces protéines agissent également lors de la réplication virale.
Alors qu’elles ne représentent qu’un tiers des protéines humaines, les protéines membranaires sont ciblées par environ 60 % des médicaments. On connaît pourtant la structure de seulement quelques centaines de ces protéines, contre une centaine de milliers de protéines globulaires.
Or ces structures aident à orienter la recherche de médicaments vers les molécules qui peuvent s’associer aux protéines correspondantes. Sans cela, trouver des composés actifs reviendrait à chercher une aiguille dans une botte de foin.
Quel problème avez-vous relevé dans l’étude de ces structures ?
Nous sommes revenus sur une publication concernant la protéine p7, impliquée dans la réplication du virus de l’hépatite C. La structure découverte par des collègues d’Harvard allait à l’encontre du consensus. On ne peut malheureusement pas observer les structures in situ tant que les protéines sont dans les membranes. Un détergent est alors utilisé pour les extraire, mais, selon sa force, il peut altérer au passage l’architecture de la protéine et conduire à une structure qui n’a aucun sens physiologique : toute la recherche basée sur ces résultats se fait dans le vide.
Comment avez-vous résolu cette controverse ?
Avec des simulations de la dynamique moléculaire, menées dans le cadre du laboratoire international associé SML1, nous avons démontré que la structure de p7 proposée par les chercheurs de Harvard était un artéfact. Parallèlement, nos collègues d’Oxford ont utilisé la RMN pour revisiter les expériences de Harvard, en prouvant que la structure précédente avait bien été altérée par le détergent. La synergie théorie/expérience a permis non seulement de constater le problème, mais aussi de l’expliquer.
La connaissance de la vraie structure de cette protéine aide à imaginer comment elle s’auto-organise pour former un canal transmembranaire, et ouvre ainsi la voie au design rationnel de médicaments ciblant la réplication du virus de l’hépatite C.
Référence
Benjamin P Oestringer, Juan H Bolivar, Mario Hensen, Jolyon K Claridge, Chris Chipot, François Dehez, Nicole Holzmann, Nicole Zitzmann, Jason R Schnell.
Re-evaluating the p7 viroporin structure
Brief Communications Arising, Nature – Octobre 2018
DOI: 10.1038/s41586-018-0561-9