
L’intelligence artificielle au service de la mécanochimie
Conduire des réactions chimiques sans solvants et avec moins de déchets, c’est la promesse de la mécanochimie. Des scientifiques de l’Institut des biomolécules Max Mousseron et de l’institut Charles Gerhardt Montpellier proposent d’utiliser des méthodes d’optimisation par intelligence artificielle pour accélérer la mise au point et le déploiement de ces réactions activées par de l’énergie mécanique.
La chimie s’est construite autour d’un geste presque immuable : dissoudre des réactifs dans un solvant et les mélanger pour les faire réagir, souvent en les chauffant. Mais une autre voie s’ouvre aujourd’hui, plus sobre et plus directe : la mécanochimie. Ici, les réactions sont déclenchées sans solvant ou presque par une action mécanique, qu’il s’agisse d’un simple broyage dans un moulin à billes ou d’intenses forces de cisaillement dans une extrudeuse à vis. Ce changement de paradigme n’est pas anodin : il permet de réduire drastiquement l’usage de substances souvent toxiques, de limiter les déchets et effluents et de simplifier les procédés. Encore faut-il apprendre à maîtriser et optimiser ces réactions solides, dont les paramètres interagissent de manière complexe : temps de broyage, proportion des réactifs et conditions physiques pour le broyeur à bille ou température, vitesse de rotation, temps de séjour ou longueur des vis pour l'extrusion. L’exploration de l’effet sur la réaction de chacun de ces paramètres pris individuellement nécessite un grand nombre d'expériences coûteuses en temps et en ressources.
Dans ce contexte, des chimistes de l’Institut des biomolécules Max Mousseron et de l’institut Charles Gerhardt Montpellier (CNRS/ENSC Montpellier/Université de Montpellier) proposent une solution originale qui convoque les outils de l’intelligence artificielle pour accélérer l’exploration de l’espace paramétrique de la mécanochimie. Leur approche, basée sur un algorithme d'optimisation bayésienne, est illustrée dans le cadre de la mise au point de réactions centrales en chimie organique et médicinale : l’amidation, ou formation d’une fonction amide à partir d’une amine et d’un acide.
L’optimisation bayésienne est une approche probabiliste qui modélise le résultat de réactions par une approche « boîte noire ». Une intégration de la variance directement dans l’optimisation permet non seulement d’atteindre le meilleur résultat, mais aussi celui qui présente la meilleure reproductibilité. Cette approche prend en compte les différents paramètres de façon holistique plutôt qu’individuellement. La force de cette démarche est de pouvoir intégrer divers objectifs comme l’optimisation des rendements, tout en minimisant l’excès de réactifs, par exemple. L’algorithme découvre progressivement l’impact et les synergies entre les différents paramètres de réaction, ainsi que les compromis à considérer entre les divers objectifs. Le choix d’un objectif intégrant un facteur environnemental permet a priori une optimisation en accord avec une démarche de synthèse priorisant la réduction de déchets. Après une phase d'apprentissage basée sur des données de synthèse conduites à paramètres variables, l'algorithme converge rapidement en proposant des conditions optimisées. Par la suite, toute optimisation supplémentaire (sur un autre composé de départ par exemple) ne nécessite qu’un nombre réduit d’expériences, permettant une individualisation des conditions de réactions pour chaque cas, ce qui est très différent d’une approche classique privilégiant des conditions supposées universelles.
L’équipe démontre ainsi l’optimisation bayésienne de la mécanosynthèse par broyage d’un synthon nécessaire à la préparation d’une famille de biopolymères d’intérêt biomédical. Cette réaction d’acylation d’un acide aminé a pu être optimisée par algorithme de manière rapide et efficace, surtout en comparaison soit avec une approche « un paramètre à la fois », soit en utilisant un plan d’expériences. La puissance de cette approche a aussi pu être démontrée pour l’optimisation bayésienne de la synthèse par extrusion réactive à haute température d’un médicament. L’antidépresseur moclobémide a pu être préparé en continu sans solvant organique, de la synthèse à la purification, générant très peu de déchets (300 g pour 1 kg de moclobémide produit).
La performance de cette approche ouvre des perspectives nouvelles pour une chimie plus durable et plus intelligente. En combinant mécanochimie et optimisation assistée par algorithmes, il devient possible d’ajuster finement les conditions pour chaque molécule, de limiter les ressources engagées et même d’anticiper les compromis entre efficacité et impact environnemental. A mesure que ces méthodes s’étendent à d’autres réactions et intègrent de nouveaux paramètres, elles dessinent les contours d’une chimie où l’expérimentation, loin d’être abandonnée, devient plus ciblée, plus sobre et profondément augmentée par le calcul. De quoi dessiner un bel avenir à la mécanochimie, grande contributrice au développement durable.
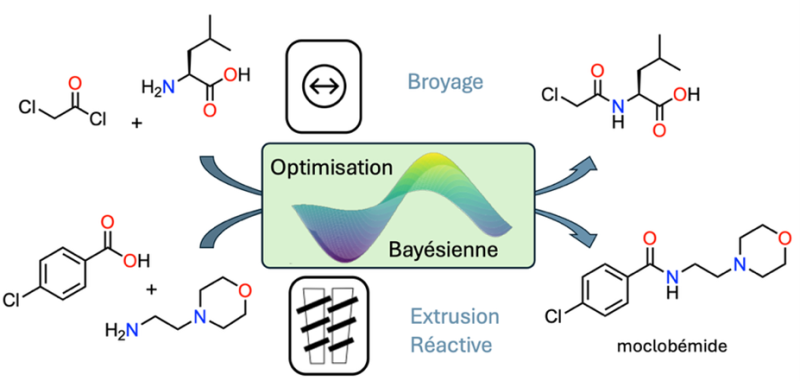
Figure : Deux réactions clés de la chimie ont été conduites par mécanochimie optimisée à l’aide d’outils d’intelligence artificielle : l’acylation d’un acide aminé pour former une brique de base importante de biopolymères (haut) et la formation d’une fonction amide pour produire le moclobémide (bas).
Références
Smart mechanochemistry: optimizing amino acid acylation with one a factor at a time, design of experiments and machine learning methods
Gallego, A.; Lavayssiere, M.; Bantreil, X.; Pétry, N.; Pinaud, J.; Giani, O.; Lamaty, F.
RSC Mechanochem 2026
DOI: 10.1039/D5MR00096C
Bayesian Optimization of Solvent-free Thermal Amidation via Reactive Extrusion
Lavayssiere, M.; Bantreil, X.; Lamaty, F.
Chem. Eur. J. 2026
https://doi.org/10.1002/chem.70914