Simulations numériques : quand voir flou permet de voir plus loin
L’étude de nombreux problèmes scientifiques actuels repose sur les simulations moléculaires qui permettent de réaliser sur ordinateur des expériences impossibles à mener en laboratoire. Qu’il s’agisse de comprendre les transformations physiques et chimiques dans les liquides ou les matériaux, ou encore les changements de conformation des protéines en biologie, la complexité des systèmes étudiés ne permet pas encore de prédire directement leur évolution sur les échelles de temps associées à ces phénomènes physiques. Un nouveau pas vient d’être franchi par une équipe pluridisciplinaire de chimistes, physiciens et mathématiciens, qui pourrait bien rendre ces phénomènes simulables. Ces résultats sont à retrouver dans la revue PNAS.
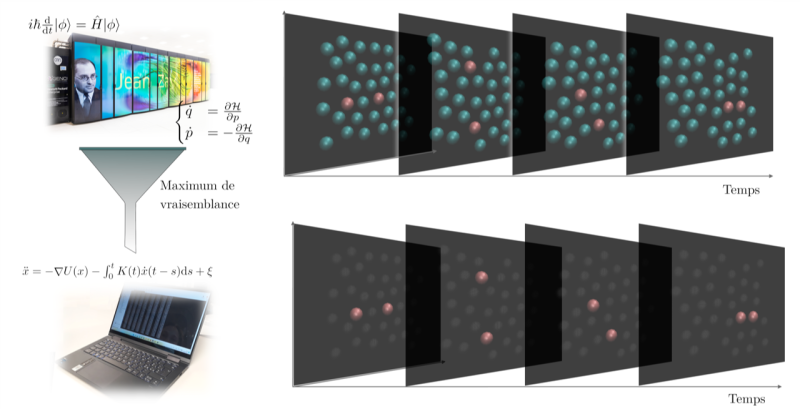
A l’échelle de la science, la simulation numérique est récente : quelques décennies seulement. Ni « expérience » ni « théorie » au sens propre de type « papier/crayon », les simulations moléculaires sur ordinateur et supercalculateurs sont pourtant devenues en peu de temps un maillon essentiel de la recherche en chimie. Aujourd’hui, elles permettent déjà, en suivant la trajectoire de chaque atome, de prédire l’évolution de petits systèmes de quelques atomes à quelques milliers d’atomes durant quelques picosecondes à quelques nanosecondes, selon la finesse de la description adoptée. Un tour de force quand on connaît le nombre de variables qui entrent en jeu et la taille des calculs associés à leur modélisation. Mais voilà : simuler un phénomène aussi simple que la dissolution d’un sel en ions dans un solvant reste une tâche bien trop complexe qui prendrait des mois et ne donnerait pas forcément un résultat fiable. On doit pour cela simplifier la description du système et quitter l’échelle de l’atome en regardant les choses « d’un peu plus loin ». Dé-zoomer peut se faire en introduisant un nombre réduit de variables collectives pertinentes, par exemple des distances entre certains atomes ou des angles entre certaines liaisons dans le cas des changements de conformation, ou encore une mesure de l'ordre local autour des atomes pour un changement d’état comme l’évaporation ou la cristallisation. Le coût de simulation associé à une telle description dite « à gros grains » est considérablement réduit par rapport à une simulation de l’évolution de chaque atome ou molécule, ce qui permet d'atteindre les échelles de temps nécessaires à l'étude des processus dynamiques physiques, chimiques ou biologiques. Tout l’enjeu pour le chimiste théoricien est de choisir la représentation du système la plus vraisemblable possible.
Enjeu tellement important qu’il a récemment suscité une collaboration interdisciplinaire entre chimistes, physiciens et mathématiciens du CNRS et de Sorbonne Université, baptisée MAESTRO.* Pour simplifier les systèmes théoriques étudiés à l’aide de variables collectives, les théoriciens ont développé une approche systématique qui permet de sélectionner la ou les variables représentant au mieux le système en évolution. Pour cela, il a d’abord fallu déterminer les lois physiques qui gouvernent l'évolution de chaque variable au cours du temps et les traduire par des équations mathématiques correctes. Tâche complexe puisqu’il faut y introduire un effet de mémoire lié au fait que les atomes ou les molécules interagissent avec un environnement complexe et que l'évolution future d’un système dynamique dépend de façon subtile de son passé. Ils ont ensuite proposé un outil mathématique reposant sur un algorithme de maximisation de la vraisemblance qui permet d'identifier les meilleurs modèles dynamiques pour les variables collectives sélectionnées, afin de révéler celles qui sont les plus pertinentes pour les transformations étudiées. Validée comme preuve de concept sur des systèmes modèles, cette nouvelle méthode, décrite dans la revue PNAS, est une avancée considérable en chimie théorique. Elle devrait ouvrir la voie à une meilleure compréhension des mécanismes physiques et chimiques de nombreux phénomènes complexes comme le vieillissement des batteries électriques ou le développement de catalyseurs plus performants.
* MAterials for Energy through STochastic sampling and high peRformance cOmputing, une équipe-projet de l'Institut des Sciences du Calcul et des Données de l'Alliance Sorbonne Université (https://iscd.sorbonne-universite.fr/research/sponsored-junior-teams/maestro-2)
Rédacteur: AVR
Référence
Likelihood-based non-Markovian models from molecular dynamics
Hadrien Vroylandt, Ludovic Goudenège, Pierre Monmarché, Fabio Pietrucci et Benjamin Rotenberg PNAS 29 mars 2022.